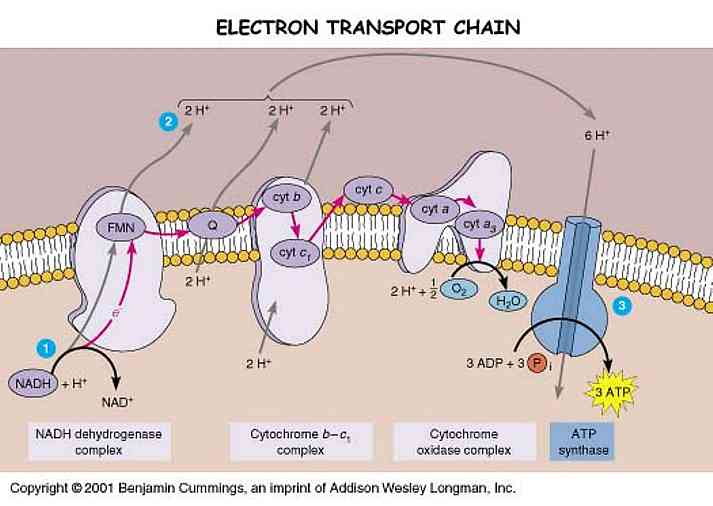
Many Parkinson's patients take Coenzyme Q10 supplements. As mentioned in the previous post, CoQ10 is part of the Electron Transport Chain -- a very important part, in fact, as it alleviates pressure on our precarious and susceptible-to-aging Complex I.
While many theorize that Complex I is shut down or is deficient in PD (1, 2, 3, 4), others believe that deficient activity of the CoQ10 pool beside Complex I is more to blame (5, 6). The CoQ10 theory claims that PD causes a deficiency in the CoQ10 pool that carries electrons from Complex I to their next destination without producing ROS. As a result of low CoQ10, electrons build up in Complex I and get released from the entrance because they cannot leave through the exit.

Some PD patients are able to take CoQ10 supplements and improve their condition (7).
It is my opinion that CoQ10 is a palliative treatment and not a long-term solution. The Ndi1gene therapy discussed in the previous post is a better option if it makes it to, and proves robust in clinical trials. My reasoning is that a genetic replacement for Complex I is a more stable therapy than a persistent aid to CoQ10: it is more permanent and a more widespread solution; a large portion of PD patients do not have CoQ10 deficiencies. Ndi1 would also contribute to the sustaining of the proton gradient in the mitochondria, also vital to creating energy in the ETC.

Greenamyre, J. (2001). Response: Parkinson's disease, pesticides and mitochondrial dysfunction Trends in Neurosciences, 24 (5) DOI: 10.1016/S0166-2236(00)01788-4
Schapira AH (1994). Evidence for mitochondrial dysfunction in Parkinson's disease--a critical appraisal. Movement disorders : official journal of the Movement Disorder Society, 9 (2), 125-38 PMID: 8196673
Morais, V., Verstreken, P., Roethig, A., Smet, J., Snellinx, A., Vanbrabant, M., Haddad, D., Frezza, C., Mandemakers, W., Vogt-Weisenhorn, D., Van Coster, R., Wurst, W., Scorrano, L., & De Strooper, B. (2009). Parkinson's disease mutations in PINK1 result in decreased Complex I activity and deficient synaptic function EMBO Molecular Medicine, 1 (2), 99-111 DOI: 10.1002/emmm.200900006
Storch, A., Jost, W., Vieregge, P., Spiegel, J., Greulich, W., Durner, J., Muller, T., Kupsch, A., Henningsen, H., Oertel, W., Fuchs, G., Kuhn, W., Niklowitz, P., Koch, R., Herting, B., Reichmann, H., & , . (2007). Randomized, Double-blind, Placebo-Controlled Trial on Symptomatic Effects of Coenzyme Q10 in Parkinson Disease Archives of Neurology, 64 (7), 938-944 DOI: 10.1001/archneur.64.7.nct60005
Greenamyre, J. (2001). Response: Parkinson's disease, pesticides and mitochondrial dysfunction Trends in Neurosciences, 24 (5) DOI: 10.1016/S0166-2236(00)01788-4
Schapira AH (1994). Evidence for mitochondrial dysfunction in Parkinson's disease--a critical appraisal. Movement disorders : official journal of the Movement Disorder Society, 9 (2), 125-38 PMID: 8196673
Morais, V., Verstreken, P., Roethig, A., Smet, J., Snellinx, A., Vanbrabant, M., Haddad, D., Frezza, C., Mandemakers, W., Vogt-Weisenhorn, D., Van Coster, R., Wurst, W., Scorrano, L., & De Strooper, B. (2009). Parkinson's disease mutations in PINK1 result in decreased Complex I activity and deficient synaptic function EMBO Molecular Medicine, 1 (2), 99-111 DOI: 10.1002/emmm.200900006
Storch, A., Jost, W., Vieregge, P., Spiegel, J., Greulich, W., Durner, J., Muller, T., Kupsch, A., Henningsen, H., Oertel, W., Fuchs, G., Kuhn, W., Niklowitz, P., Koch, R., Herting, B., Reichmann, H., & , . (2007). Randomized, Double-blind, Placebo-Controlled Trial on Symptomatic Effects of Coenzyme Q10 in Parkinson Disease Archives of Neurology, 64 (7), 938-944 DOI: 10.1001/archneur.64.7.nct60005